AHP IST EINE FAMILIE
VON SELTENEN UND STARK BEEINTRÄCHTIGENDEN
GENETISCHEN ERKRANKUNGEN
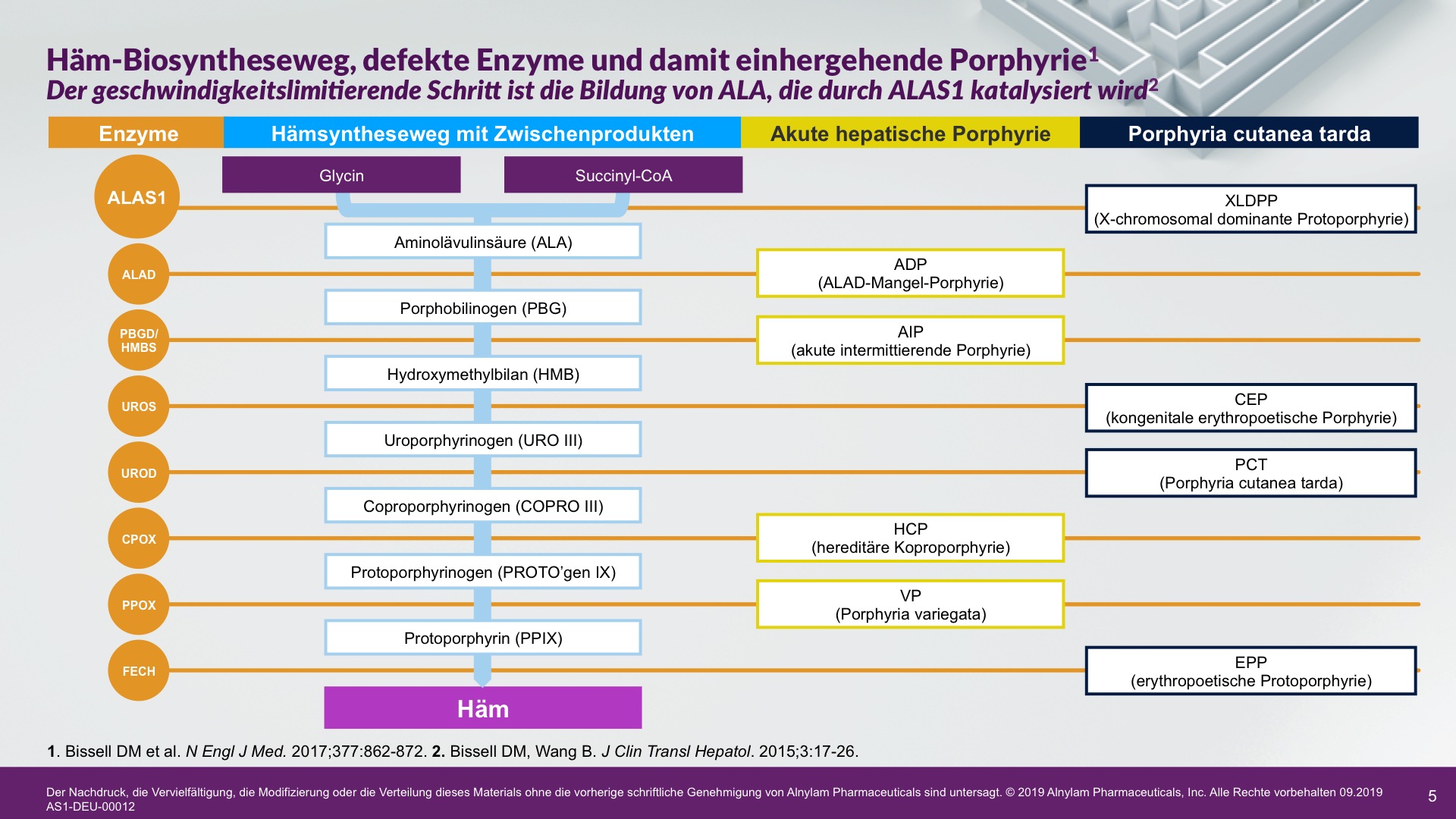
Jeder Porphyrie-Subtyp geht mit einem Enzymdefekt im Häm-Biosyntheseweg einher. Sie werden wie folgt klassifiziert: akute hepatische Porphyrie (AHP) – die als plötzliche akute neuroviszerale Attacke auftreten kann – und Porphyria cutanea tarda.1-3
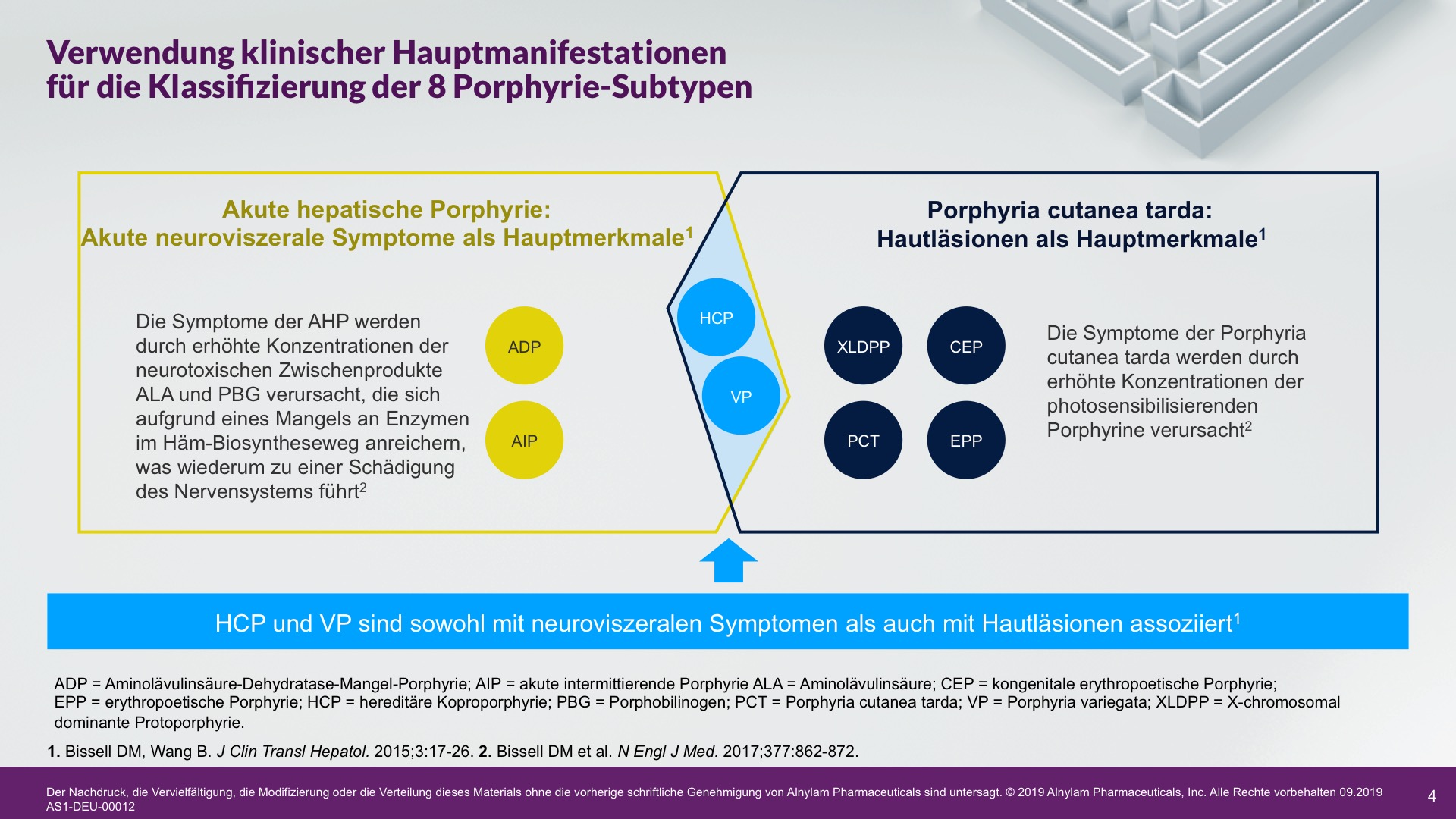
Verwendung klinischer Hauptmanifestationen für die Klassifizierung in 8 Porphyrie-Subtypen3,4


ADP=Aminolävulinsäure-Dehydratase-Mangel-Porphyrie; AIP=akute intermittierende Porphyrie; ALA=Aminolävulinsäure; CEP=kongenitale erythropoetische Porphyrie; EPP = erythropoetische Porphyrie; HCP = hereditäre Koproporphyrie; PBG=Porphobilinogen; PCT=Porphyria cutanea tarda; PV=Porphyria variegata; XLDPP=X-chromosomal dominante Protoporphyrie.
URSACHEN DER AHP
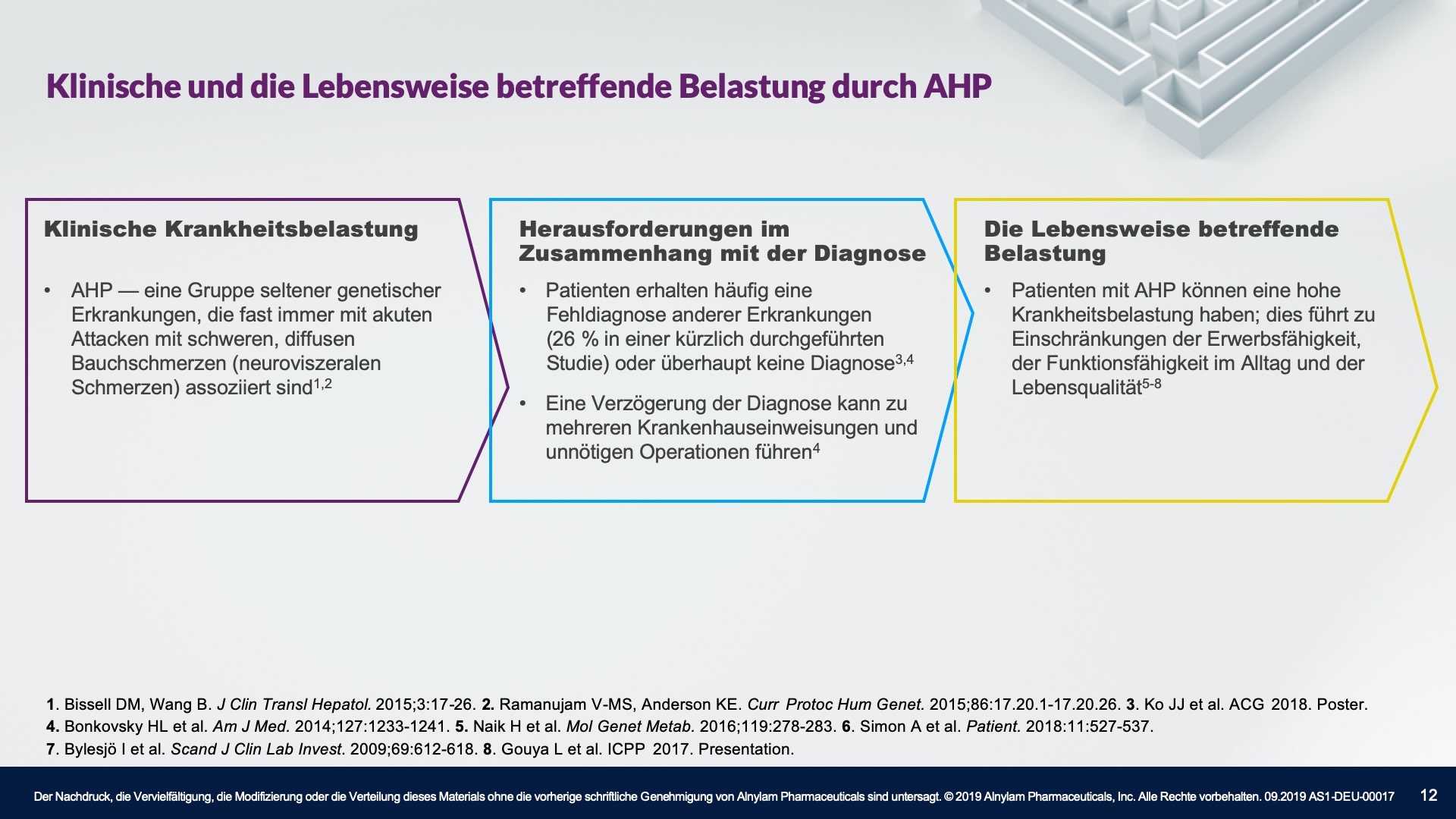
Die Pathophysiologie der Porphyrie umfasst 8 genetische Stoffwechselerkrankungen des Häm-Biosynthesewegs. AHP, ein Subtyp der Porphyrie, manifestiert sich häufig als plötzliche akute Attacken, die eine Krankenhauseinweisung erfordern und durch überschüssige(s) ALA und PBG verursacht werden.2-5


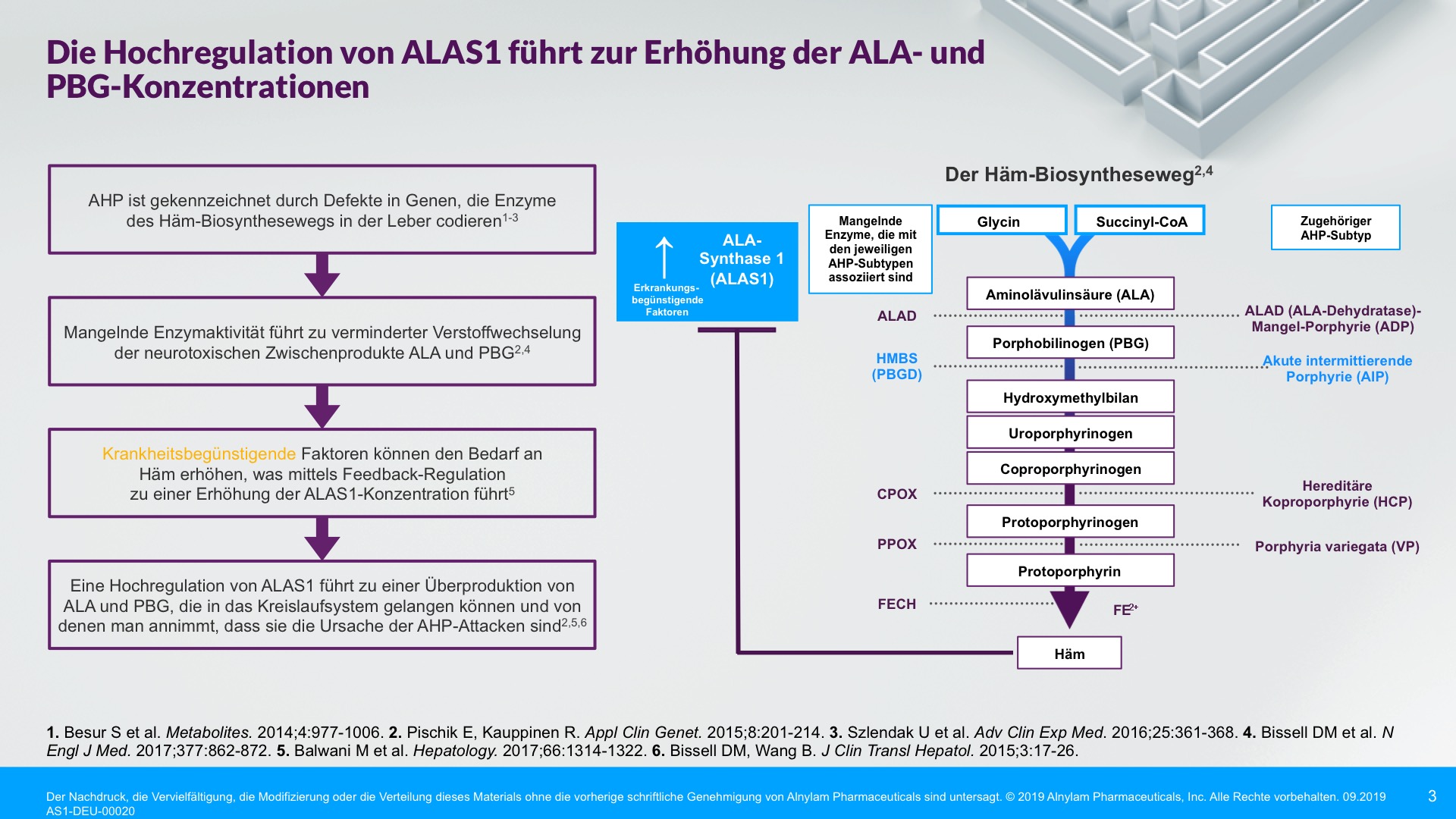
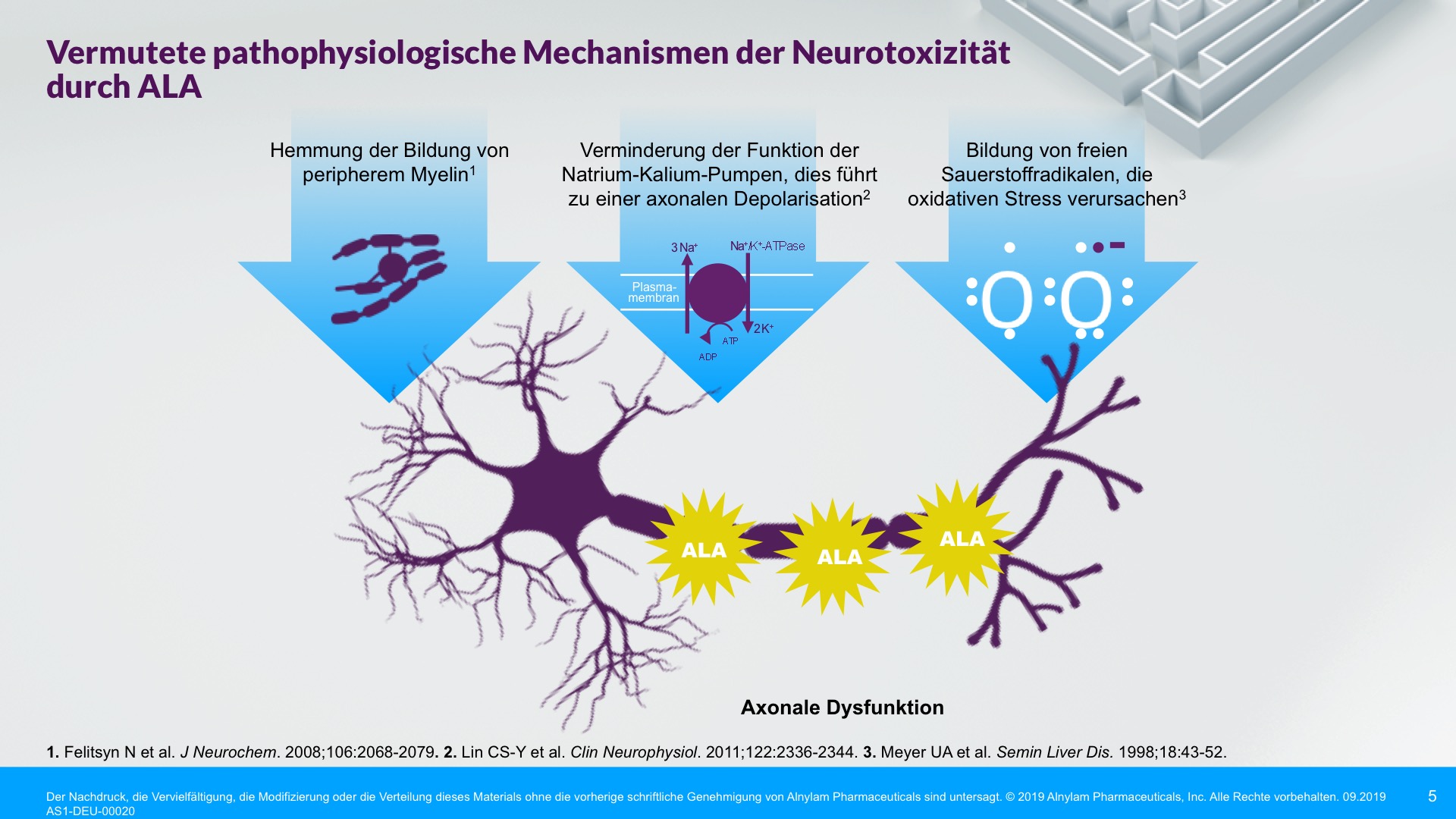
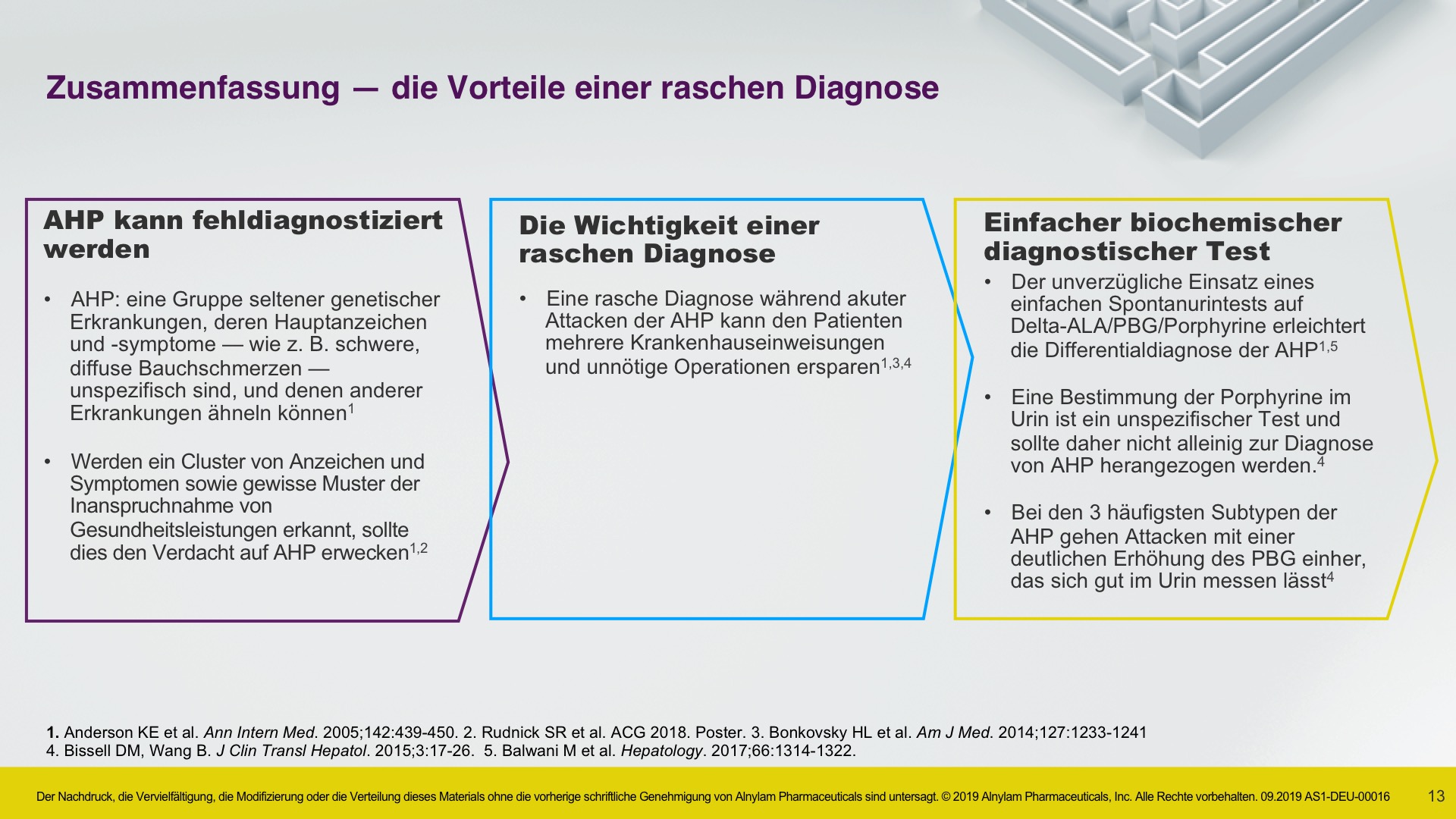
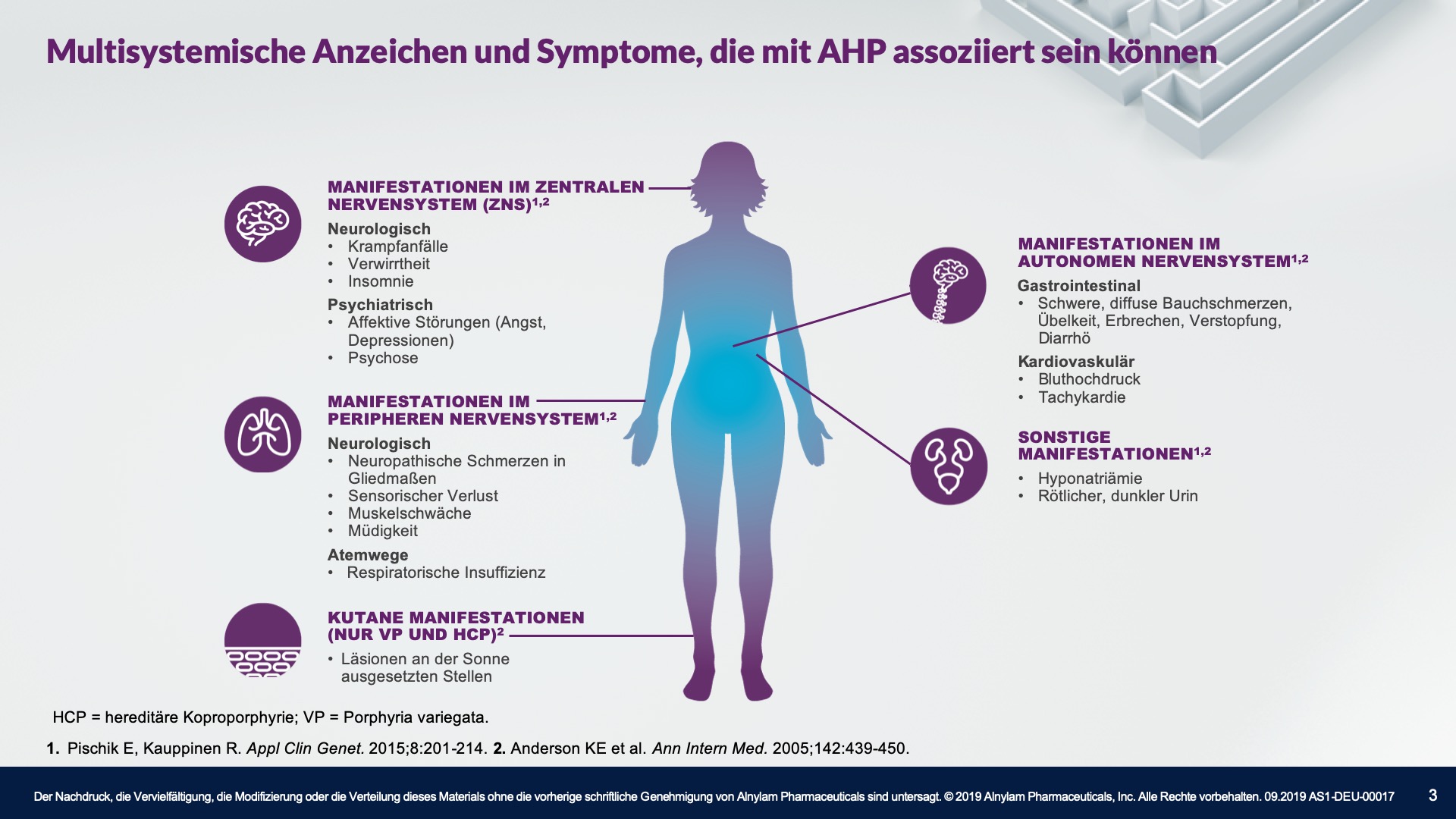
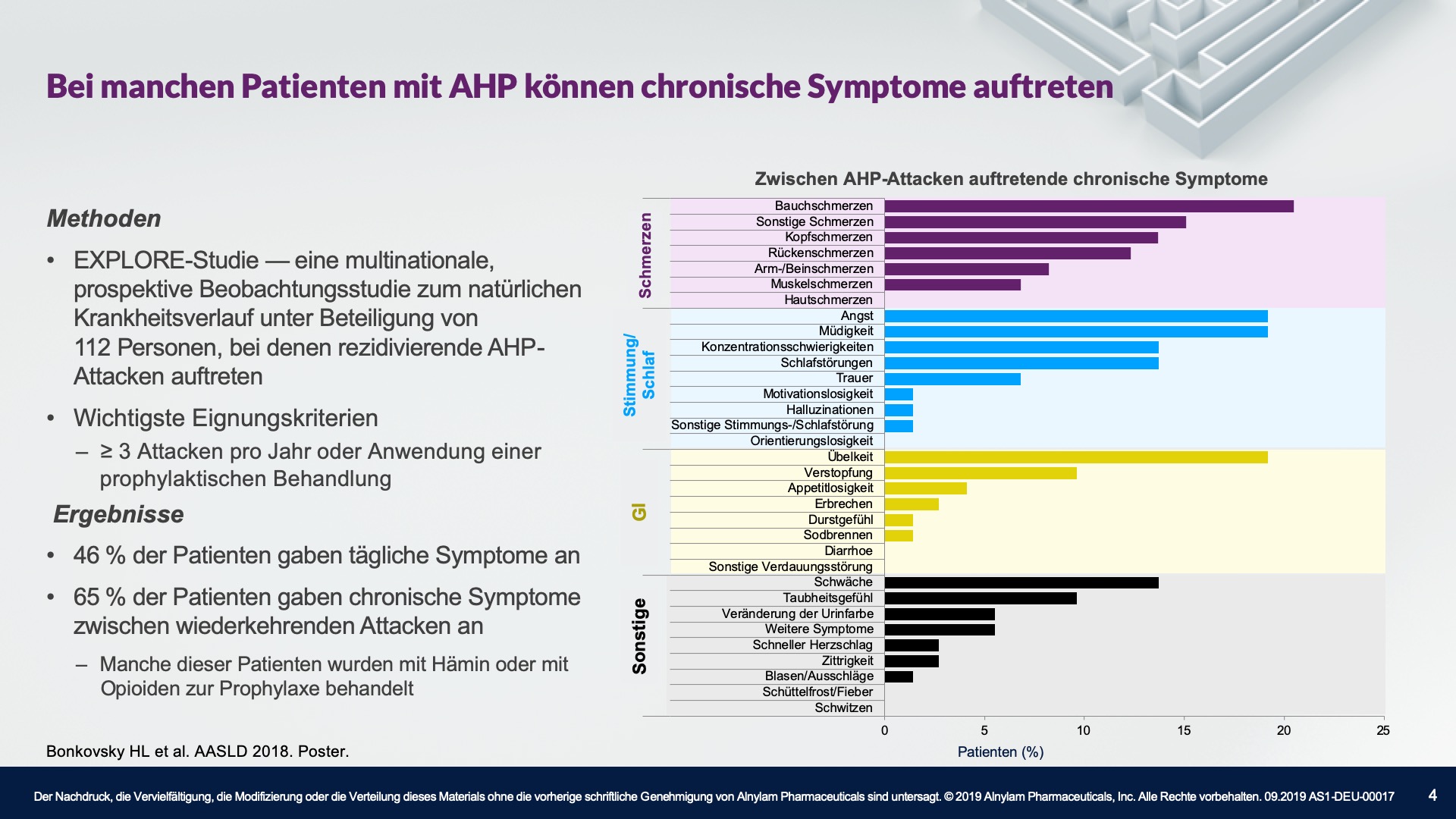
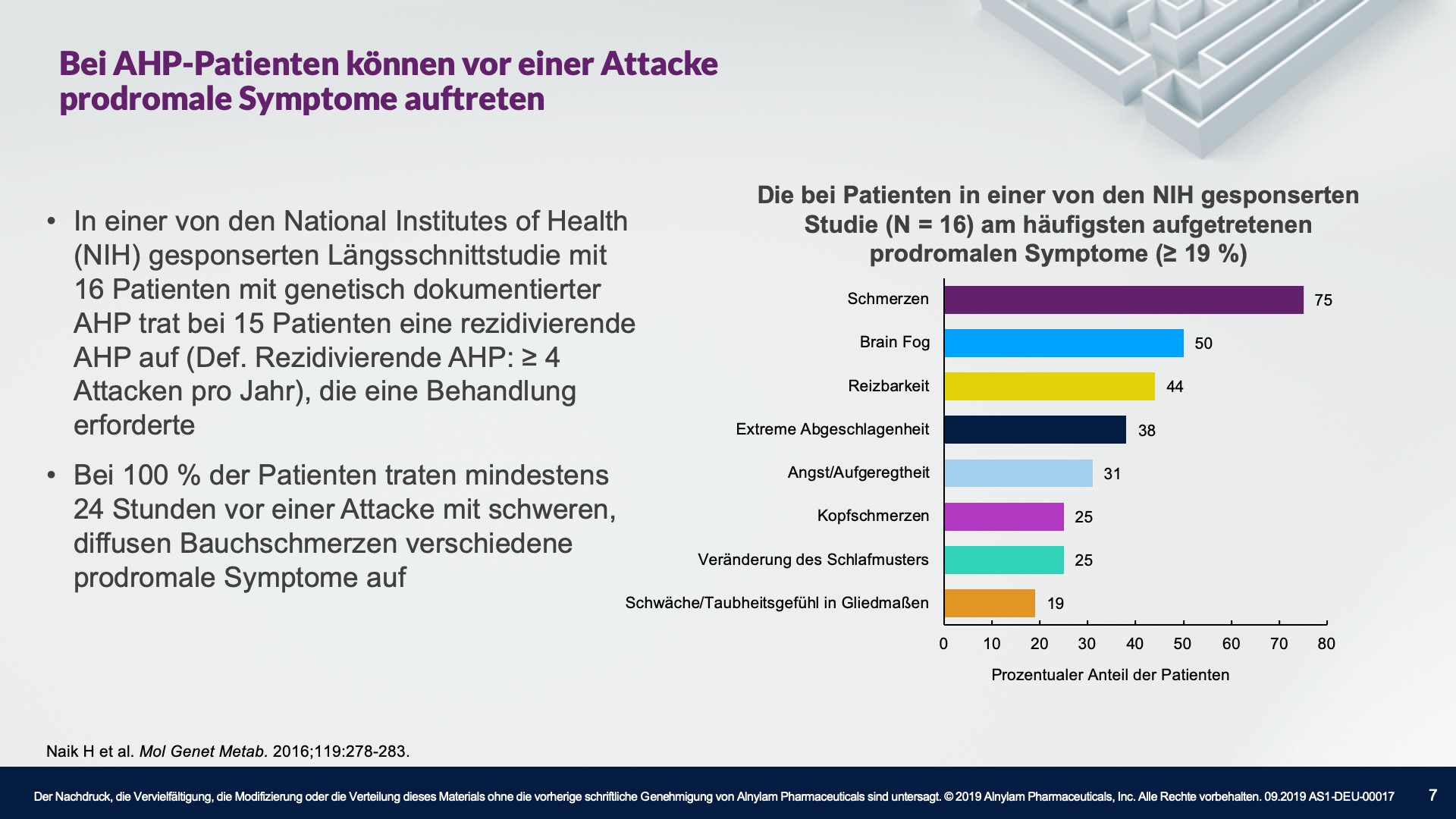
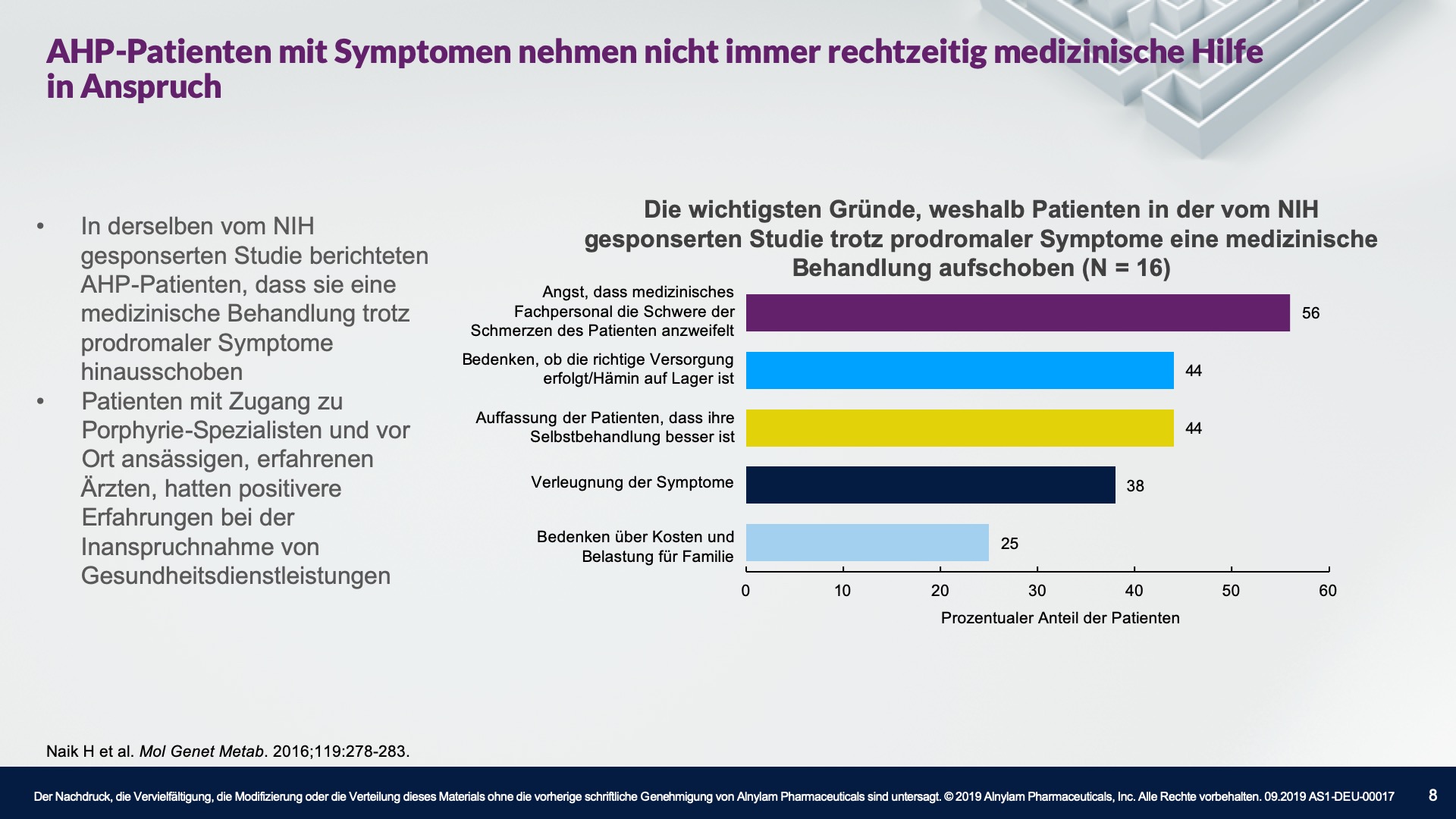
AHP wird durch Enzymdefekte verursacht, die zur Anreicherung neurotoxischer Zwischenprodukte in der Leber führen — Aminolävulinsäure (ALA) und Porphobilinogen (PBG). Die Hochregulierung von ALAS 1 (Aminolävulinsäure-Synthase-1) ist ein zentraler pathophysiologischer Mechanismus, der der AHP zugrunde liegt und zu dieser Anreicherung neurotoxischer Zwischenprodukte führt. ALA und PBG gelangen anschließend ins Kreislaufsystem, und es wird davon ausgegangen, dass sie Nervenläsionen verursachen, die zu Funktionsstörungen im autonomen, zentralen und peripheren Nervensystem führen. Eine Funktionsstörung in diesen Systemen kann bei Patienten zu schweren, diffusen Bauchschmerzen sowie zu einem oder mehreren der folgenden Symptome führen: Angst, Verwirrtheit, Übelkeit, Erbrechen, Gliederschwäche oder -schmerzen. Dies kann zu irreversiblen Neuropathien und langfristigen Krankheitskomplikationen führen, wie z. B. Nierenerkrankungen und hepatozellulärem Karzinom. Ein erhöhter PBG-Wert ist ein wichtiger hochspezifischer diagnostischer Marker. Es wird angenommen, dass ALA das primäre Neurotoxin ist, das für die akuten Attacken, die anhaltenden Symptome zwischen Attacken und die lang andauernde oder möglicherweise permanente Neuropathie bei AHP verantwortlich ist. Akute Attacken dauern im Allgemeinen 3 bis 7 Tage, aber die Erholungsphase kann lange dauern und in chronischen Restsymptomen münden.1-9
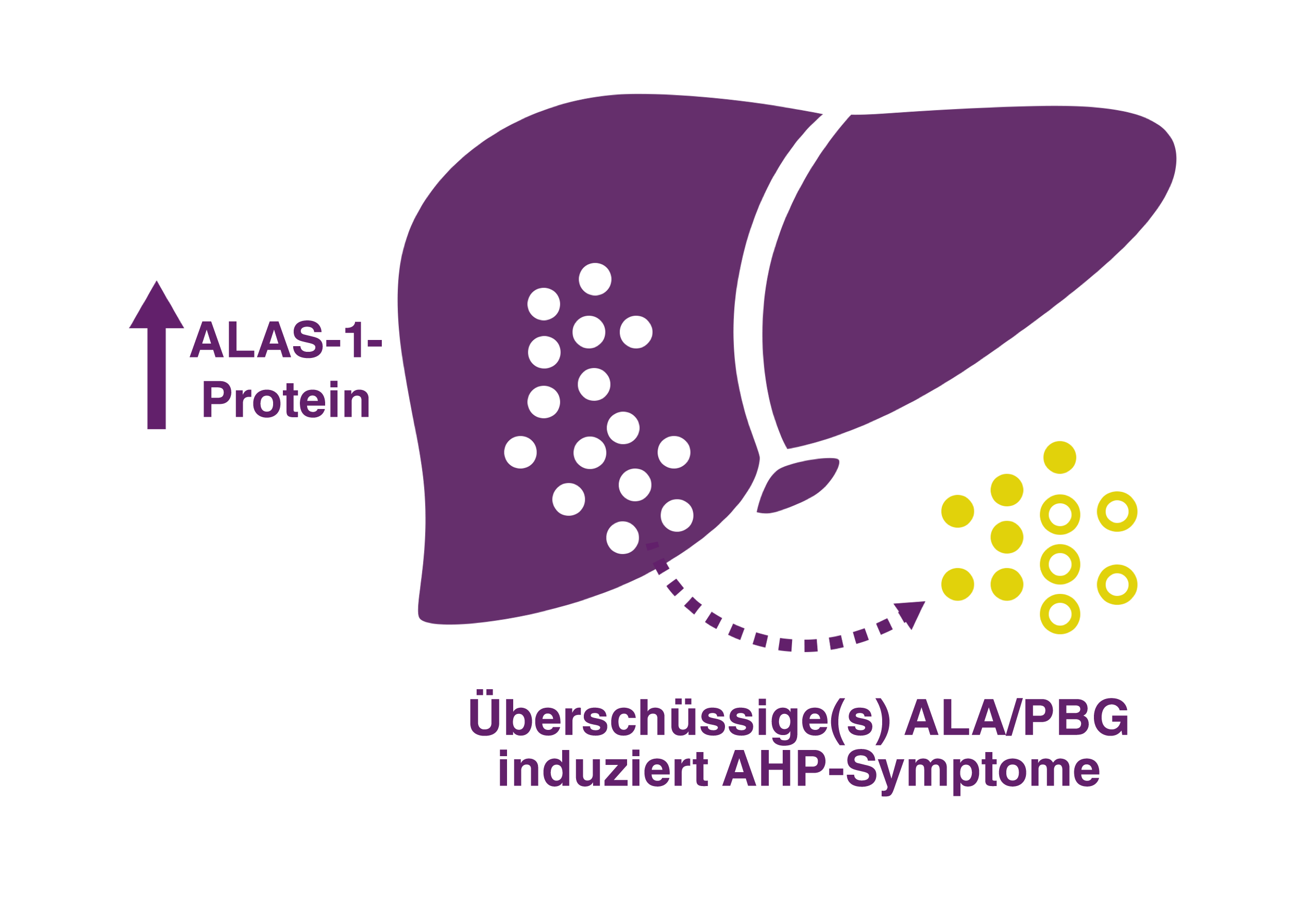
AHP UMFASST 4 SUBTYPEN
Es gibt 4 AHP-Subtypen, die von unterschiedlichen Enzymmängeln im Häm-Biosyntheseweg in der Leber herrühren. Bei ca. 80 % der Fälle handelt es sich um akute intermittierende Porphyrie (AIP), gefolgt von Porphyria variegata (PV), hereditärer Koproporphyrie (HCP) und der extrem seltenen ALA-Dehydratase-Mangel-Porphyrie (ADP). Möglicherweise wird die Prävalenz von AIP aufgrund von Schätzungen, die nur auf Patienten mit symptomatischer Erkrankung anstatt auf Patienten mit Enzymmutation basieren, als zu gering ausgewiesen.2,4,8,10,11
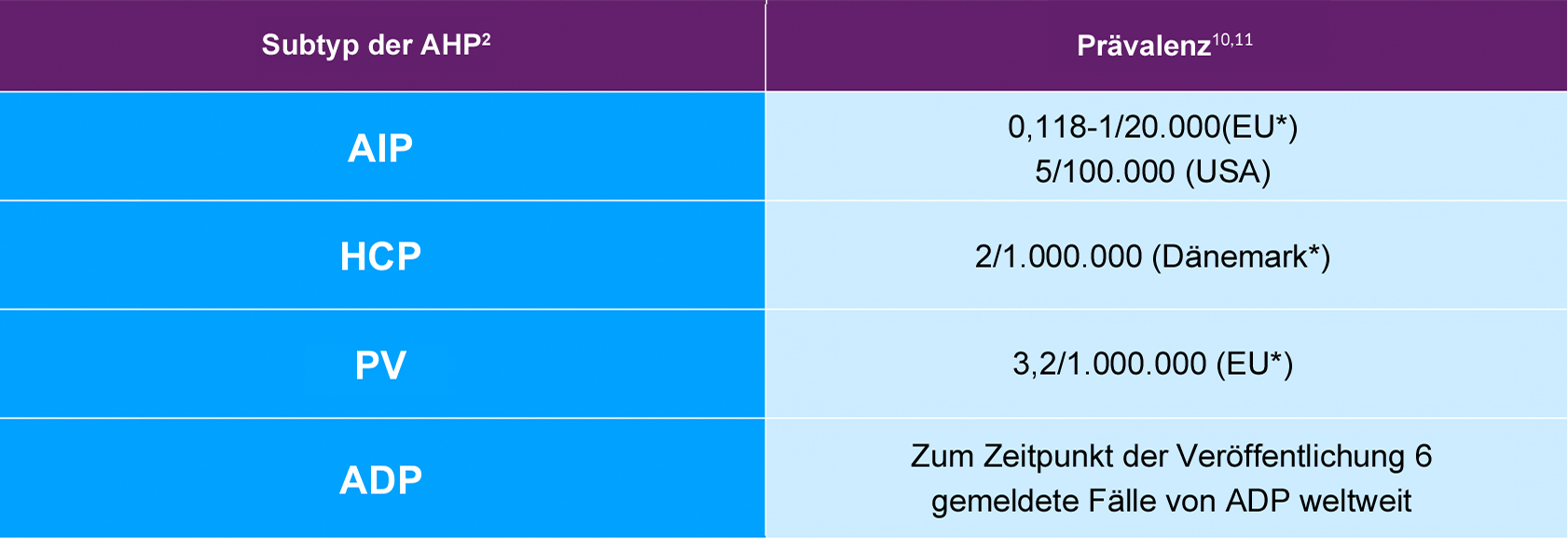
*Prävalenzdaten aus den genannten Ländern wurden aufgrund laufender Forschung und einer relativ hohen Prävalenz angeführt.
EPIDEMIOLOGIE VON AHP
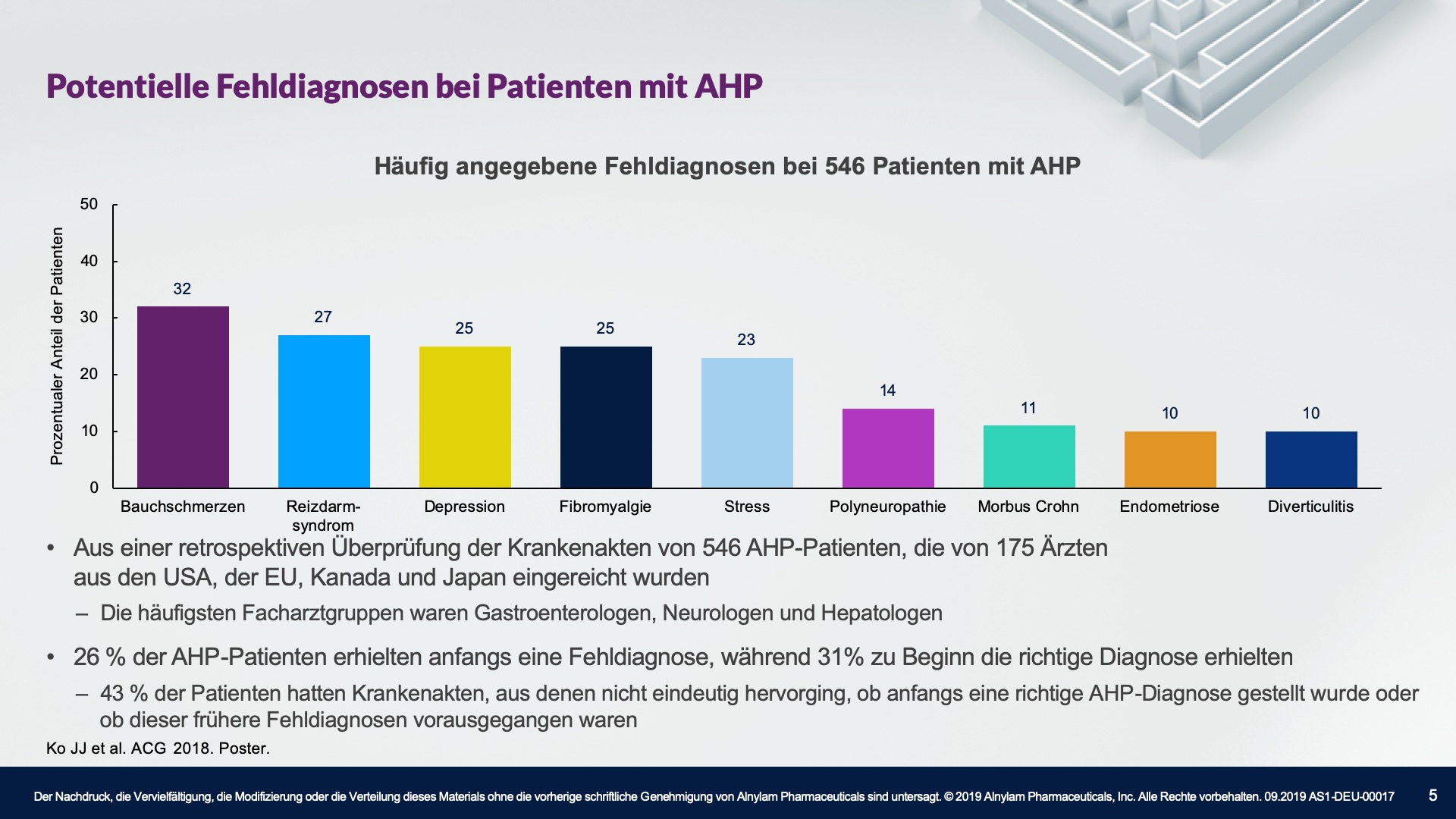
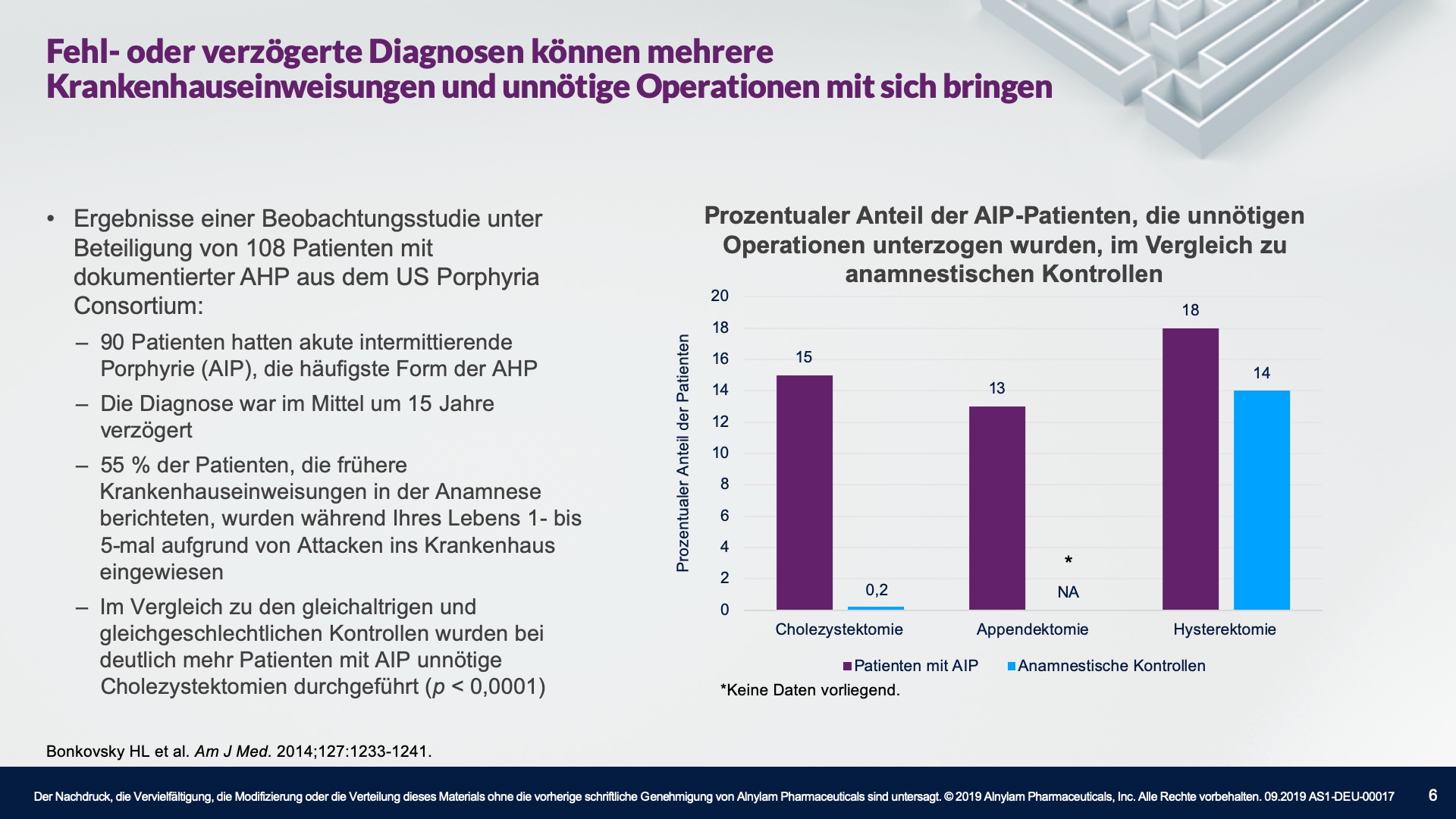
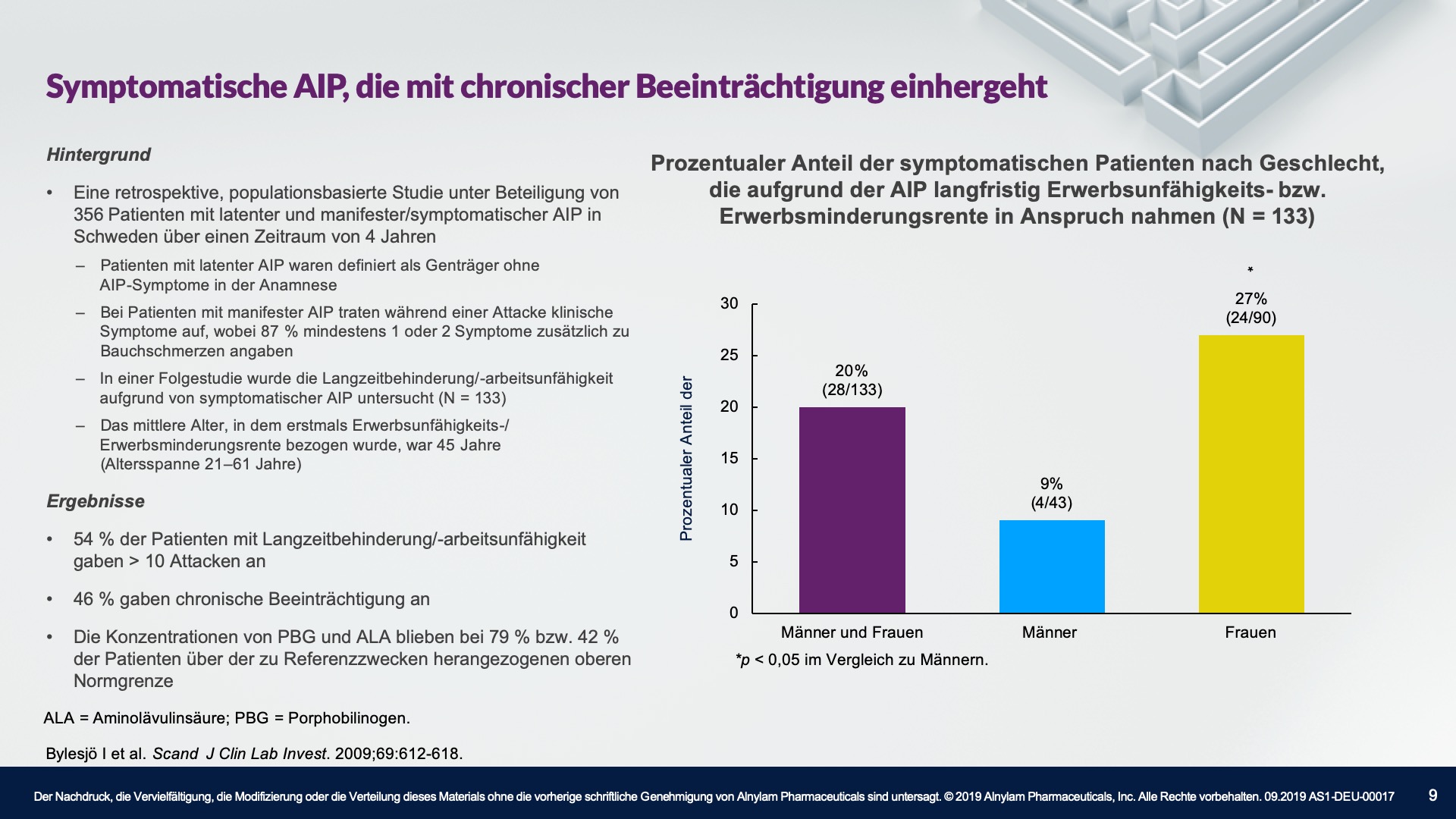
AHP betrifft Menschen aller ethnischen Zugehörigkeiten, wobei die Prävalenz unter der weißen Bevölkerung am höchsten ist. Die meisten Fälle treten bei Frauen im gebärfähigen Alter auf, wobei die meisten Attacken im Alter von 15 bis 45 Jahren auftreten. Es ist wichtig zu erwähnen, dass das Risiko langfristiger Krankheitskomplikationen, wie z. B. einer Porphyrie-bedingten Nierenerkrankung oder eines hepatozellulären Karzinoms, nach der Menopause bestehen bleibt. Bei manchen Patienten treten stark beeinträchtigende und sogar lebensbedrohliche Attacken auf.2-4,10-16
DIE MEISTEN FÄLLE TRETEN BEI FRAUEN AUF12†

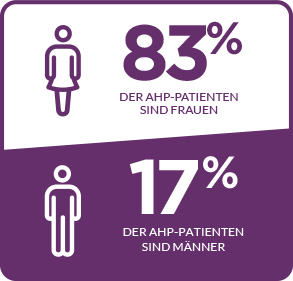
†In einer Studie unter Beteiligung von 108 Patienten.